Author: OneAngstrom
Understanding Backbone Node Attributes in Molecular Modeling
Efficiently Managing Computational Jobs in SAMSON Cloud.
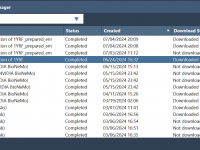
For molecular modelers, working on complex molecular dynamics or protein structure prediction tasks often involves computationally intensive calculations. Managing these tasks effectively can be a challenge, especially when dealing with cloud-based workflows. This is where the Job Manager in SAMSON…
Efficient Transition Path Optimization with P-NEB in SAMSON

Understanding File Attributes in SAMSON’s Node Specification Language
Enhancing Molecular Animations with the Play Reverse Path Feature
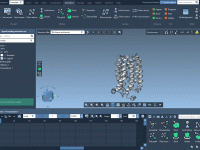
Molecular modelers often face the challenge of effectively visualizing dynamic processes like trajectories or transitions between conformations. These dynamic visualizations can be crucial for understanding molecular mechanisms and communicating findings. To help address this, SAMSON provides a simple yet powerful…