Author: OneAngstrom
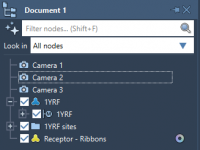
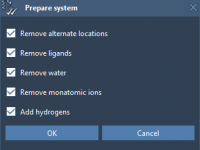
A Step-by-Step Guide to Preparing Proteins for Docking in SAMSON
Effortlessly Managing Cloud Jobs with SAMSON’s Job Manager
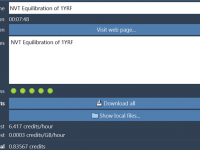
Understanding Molecule Attributes in SAMSON’s Node Specification Language
Simplifying Molecular Design: A Step-by-Step Guide to Installing SAMSON
Enhancing Molecular Motions with Real-Time Minimization in SAMSON
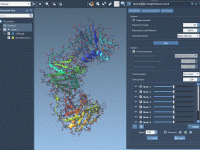
One of the most intriguing challenges molecular modelers face is simulating realistic conformational changes in biomolecular structures. Whether you're examining protein binding sites or RNA folding patterns, ensuring that structures remain stable during simulated movements can be critical to successfully…
Effortlessly Managing Molecular Simulations with the SAMSON Job Manager
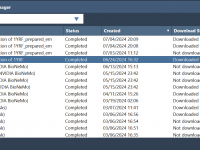
Molecular modeling professionals and researchers often grapple with efficiently organizing and managing their computational jobs. Whether it’s running molecular dynamics simulations or protein structure predictions, keeping track of these tasks can become overwhelming, especially when using cloud infrastructures. Enter the…