Mastering Protein Transition Visualization with Path Energies
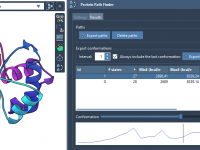
Understanding conformational transitions in proteins is a cornerstone challenge for molecular modelers, particularly when analyzing energy barriers or plotting path energies. If you’ve ever found yourself puzzled by how to visualize and interpret these transition paths, SAMSON’s Protein Path Finder…