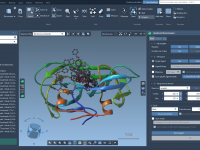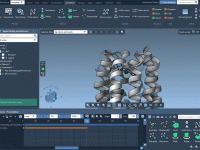
As a molecular modeler, have you ever faced the challenge of translating complex molecular structures and reactions into straightforward and captivating presentations? Whether it’s for a research conference, a classroom lecture, or simply sharing a visualization with your peers, presenting…