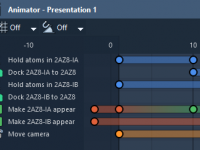
Mastering Camera Animations in SAMSON for Better Molecular Presentations
A Practical Guide to Molecule Attributes in SAMSON’s Node Specification Language
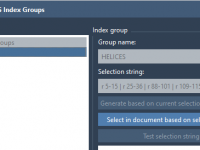
Creating Custom Index Groups for Enhanced GROMACS Simulations
Mastering Center-of-Mass Pulling with GROMACS Wizard
Center-of-mass (COM) pulling is an essential technique in molecular modeling, especially for simulating molecular interactions, exploring conformational changes, or setting up umbrella sampling studies to calculate free energy profiles. However, getting started with COM pulling simulations can be daunting due…
Practical Molecular Selection with NSL Expressions in SAMSON
If you're working on molecular modeling, one common challenge is effectively identifying specific molecular features and associations within your models. Whether it's locating binding pocket residues, identifying water molecules for mutagenesis focus, or analyzing ligand interactions, precision in selection can…