Mastering Molecular Fragment Orientation in SAMSON.
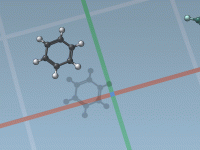
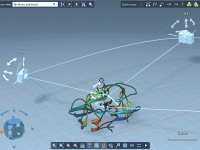
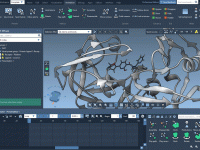
One of the most common challenges for molecular modelers is efficiently building molecular structures while maintaining realistic fragment orientations. Incorrect orientations can lead to implausible geometries or redundant refinements, consuming precious time. This challenge is particularly frustrating when constructing complex…